
 Osteosarcoma, condrosarcoma e sarcoma di Ewing sono tre termini molto simili tra loro ma che in realtà non potrebbero indicare patologie più diverse.
Osteosarcoma, condrosarcoma e sarcoma di Ewing sono tre termini molto simili tra loro ma che in realtà non potrebbero indicare patologie più diverse.
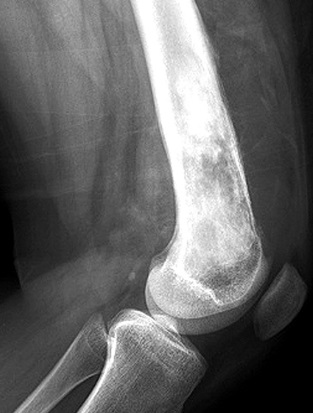
Innanzitutto l’osteosarcoma, o sarcoma osteogenico, è il tipo più frequente di cancro alle ossa che porta alla generazione di tessuto osseo immaturo all’interno di ossa in accrescimento.
Generalmente, lo si ritrova più che altro alla fine delle ossa lunghe e spesso a livello delle ginocchia; colpisce principalmente pazienti di sesso femminile con un’età inferiore ai 25 anni.
Consiste dunque in un tumore maligno: nonostante questo tipo di tumore sia molto raro, rappresenta il cancro alle ossa più comune.
Il secondo tumore più frequente alle ossa è invece il condrosarcoma che rappresenta circa un quarto dei tumori ossei maligni. Errore da non commettere consiste nel confondere questi due sarcomi.
Il condrosarcoma è infatti una neoplasia, maligna anch’essa, che porta ad un’anomala produzione di cartilagine ialina con conseguente errata formazione di tessuto osseo o cartilagineo.
I pazienti affetti da questa patologia riportano tumefazioni o protuberanze compatte in genere localizzate nelle ossa delle braccia, delle gambe o delle pelvi.
Solitamente tale neoplasia ha una prognosi più favorevole rispetto all’osteosarcoma. Infine, esiste ancora un’altra tipologia di tumore osseo maligno chiamato sarcoma di Ewing.
Nonostante la sua origine non sia ancora stata ben individuata, si ritiene che scaturisca o da una cellula embrionale o da una cellula staminale mesenchimale, capace cioè di differenziarsi in diversi tessuti.
Viene in genere definito dai patologi come un “endotelioma diffuso al tessuto osseo” simile a rari tumori dei tessuti molli. Rappresenta uno dei tumori ossei maligni più rari, colpendo circa 3 persone ogni milione sotto i 20 anni.
Il range di età più colpito, nel 90% dei pazienti, va dai 5 ai 25 anni. A differenza però dell’osteosarcoma, colpisce prevalentemente i ragazzi che presentano anche una prognosi più negativa rispetto alle ragazze.
Le ossa più colpite sono le pelvi, le ossa lunghe di gambe e braccia e le scapole.
Data la maggior frequenza di pazienti in tenera età (età media: 15 anni), tale neoplasia viene considerata come un ‘tumore pediatrico’ tra i più aggressivi: tende infatti ad essere recidiva ed a metastatizzare più dei precedenti, gli studi a riguardo sono in continuo aggiornamento.
Ad oggi, il trattamento multidisciplinare condotto in appositi centri di cura, con dottori specializzati nella terapia dei tumori ossei, permette ai pazienti di conseguire una guarigione completa in oltre il 70% dei casi.
Ovviamente, dipende tutto anche dalla tempistica e dalla prevenzione.
In linea generale il trattamento prevede tre possibili strategie: chemioterapia, radioterapia e chirurgia.
Più nel dettaglio, in pazienti affetti da osteosarcoma, la chirurgia (allotrapianto, protesi metalliche, amputazione nella peggiore delle ipotesi) rappresenta la prima scelta terapeutica, soprattutto nei casi in cui il tumore sia ben localizzato e facilmente asportabile.
Se invece si tratta di un osteosarcoma esteso può rendersi necessario un periodo di tre mesi di chemioterapia seguito dall’asportazione chirurgica e poi ancora eventualmente da radioterapia (soprattutto se la completa resezione delle cellule alterate non è possibile a causa della localizzazione tumorale).
Nei casi di condrosarcoma invece la chemio e la radioterapia non sono indicate, soprattutto per neoplasie di basso grado, ma possono essere prese in considerazione quando la prognosi si fa più critica.
La via preferenziale consiste sempre nella resezione chirurgica, la cui esecuzione dipende da molti fattori cito-istologici.
Studi recenti condotti da Bovee e Sandberg (2004-2005) hanno inoltre dimostrato che “vi è una predisposizione genetica per tale malattia e che quindi, conoscendo più a fondo i geni e le proteine coinvolte, in futuro saranno possibili trattamenti migliori”.
Infine, per quanto riguarda il sarcoma di Ewing, si ritrovano ancora le tre strategie d’intervento: chemioterapia, prima e dopo la resezione per ridurre le dimensioni della massa tumorale e per eliminare ogni possibile cellula mutata non asportata, e radioterapia quando l’asportazione non è praticabile.
In generale, “le persone trattate con questa metodologia hanno una sopravvivenza del 70-75% dopo 5 anni”, percentuale che aumenta di anno in anno anche grazie alle innovazioni in campo chemioterapico.
Essendo un tumore pediatrico, esistono oggi nuovi approcci chirurgici che permettono al medico di inserire protesi allungabili per accompagnare l’accrescimento osseo dei bambini.
Gli studi più recenti su queste protesi (Grimer, 2000) hanno riscontrato “un costante utilizzo anche a 5 anni dall’impianto nella maggior parte dei pazienti (85%)”.
I meccanismi di allungamento meccanico sono tra i più diversi ed alcuni permettono inoltre di evitare un successivo intervento chirurgico.